, . - decrease / increase font size
DEL - remove font from current set Home - restore font set to initial
Case Studies
Protein-RNA Interaction Modeling
M. Parisien (with T. Sosnick, T. Pan, and K. Freed) used Beagle to develop a first-generation algorithm for the prediction of the RNA-protein interactome.
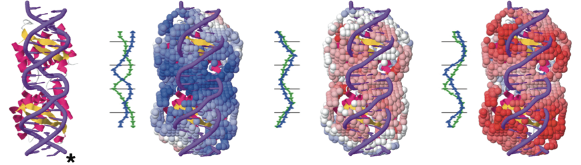
Approach. Non-coding RNAs often function in cells through specific interactions with their protein partners. Experiments alone cannot provide a complete picture of the RNA-protein interactome. To complement experimental methods, computational approaches are highly desirable. No existing method, however, can provide genome-wide predictions of docked RNA-protein complexes. the application of computational predictions, together with experimental methods, will provide a more complete understanding on cellular networks and function of RNPs. The approach makes use of a rigid-body docking algorithm and a scoring function custom- tailored for protein-tRNA interactions. Using Swift, Beagle screened about 300 proteins per day on 80 nodes of 24 cores (11% of the total XE6’s power).
Results. The scoring function can identify the native docking conformation in large sets of decoys (100,000) for many known protein-tRNA complexes (4TRA shown here). (left) Scores for true positive complexes (●)(N=28) are compared to true negative ones of low (▼)(N=40) and high (▲) (N=40) isoelectric points. (right) Because the density curve of the true positives, which have pI < 7, has minimal overlap with the curve of the low pI true negatives (blue area), the scoring function has the specificity to identify tRNA-binding proteins.